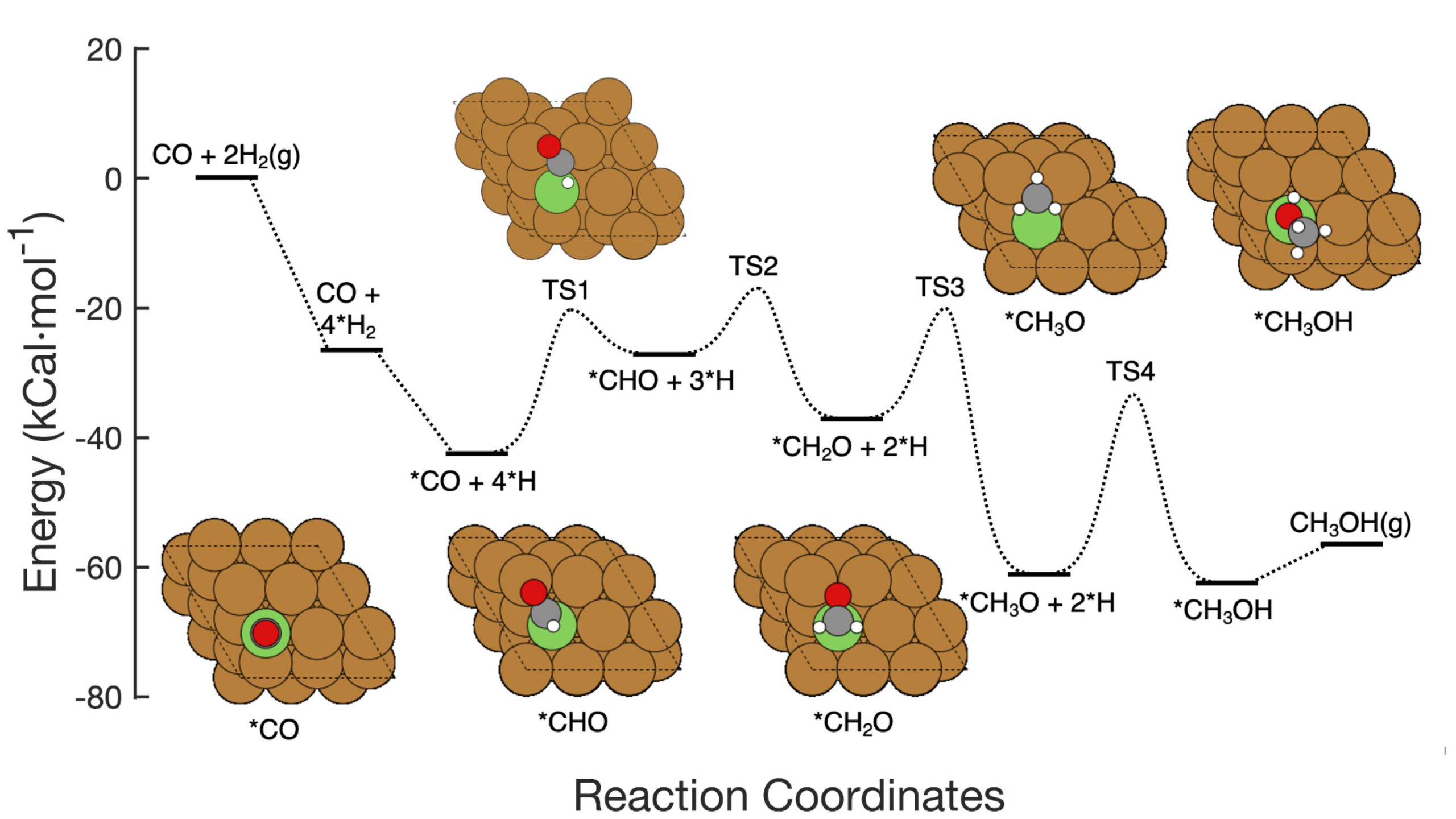

This graphic shows the seven-step reaction pathway of CO hydrogenation to methanol over copper-based catalysts, including the reactants at each step, schematic atomic arrangements of the intermediates, and the energy barriers activation required to get from step to step. The Brookhaven Lab team demonstrated a machine learning framework that successfully identified which steps/combinations of steps to tweak to improve methanol production. Their work could help guide the design of new catalysts to achieve that goal and the framework can be applied to optimize other reactions. Credit: Brookhaven National Laboratory
Chemists at the US Department of Energy’s Brookhaven National Laboratory have developed a new machine-learning (ML) framework that can zero in on which steps of a multistep chemical conversion should be tweaked to improve productivity. The approach could help guide the design of catalysts—chemical “dealmakers” that speed up reactions.
The team developed the method to analyze the conversion of carbon monoxide (CO) to methanol using a copper-based catalyst. The reaction consists of seven fairly straightforward elementary steps.
“Our goal was to identify which elementary step in the reaction network or which subset of steps controls the catalytic activity,” said Wenjie Liao, the first author on a paper describing the method just published in the journal Catalysis Science & Technology. Liao is a graduate student at Stony Brook University who has been working with scientists in the Catalysis Reactivity and Structure (CRS) group in Brookhaven Lab’s Chemistry Division.
Ping Liu, the CRS chemist who led the work, said, “We used this reaction as an example of our ML framework method, but you can put any reaction into this framework in general.”
Targeting activation energies
Picture a multistep chemical reaction as a rollercoaster with hills of different heights. The height of each hill represents the energy needed to get from one step to the next. Catalysts lower these “activation barriers” by making it easier for reactants to come together or allowing them to do so at lower temperatures or pressures. To speed up the overall reaction, a catalyst must target the step or steps that have the biggest impact.
Traditionally, scientists seeking to improve such a reaction would calculate how changing each activation barrier one at a time might affect the overall production rate. This type of analysis could identify which step was “rate-limiting” and which steps determine reaction selectivity—that is, whether the reactants proceed to the desired product or down an alternate pathway to an unwanted byproduct.

Brookhaven Lab chemist Ping Liu and Wenjie Liao, a graduate student at Stony Brook University, developed a machine learning framework to identify which chemical reaction steps could be targeted to improve reaction productivity. Credit: Brookhaven National Laboratory
But, according to Liu, “These estimates end up being very rough with a lot of errors for some groups of catalysts. That has really hurt for catalyst design and screening, which is what we are trying to do,” she said.
The new machine learning framework is designed to improve these estimates so scientists can better predict how catalysts will affect reaction mechanisms and chemical output.
“Now, instead of moving one barrier at a time we are moving all the barriers simultaneously. And we use machine learning to interpret that dataset,” said Liao.
This approach, the team said, gives much more reliable results, including about how steps in a reaction work together.
“Under reaction conditions, these steps are not isolated or separated from each other; they are all connected,” said Liu. “If you just do one step at a time, you miss a lot of information—the interactions among the elementary steps. That’s what’s been captured in this development,” she said.
Building the model
The scientists started by building a data set to train their machine learning model. The data set was based on “density functional theory” (DFT) calculations of the activation energy required to transform one arrangement of atoms to the next through the seven steps of the reaction. Then the scientists ran computer-based simulations to explore what would happen if they changed all seven activation barriers simultaneously—some going up, some going down, some individually, and some in pairs.
“The range of data we included was based on previous experience with these reactions and this catalytic system, within the interesting range of variation that is likely to give you better performance,” Liu said.
By simulating variations in 28 “descriptors”—including the activation energies for the seven steps plus pairs of steps changing two at a time—the team produced a comprehensive dataset of 500 data points. This dataset predicted how all those individual tweaks and pairs of tweaks would affect methanol production. The model then scored the 28 descriptors according to their importance in driving methanol output.
“Our model ‘learned’ from the data and identified six key descriptors that it predicts would have the most impact on production,” Liao said.
After the important descriptors were identified, the scientists retrained the ML model using just those six “active” descriptors. This improved ML model was able to predict catalytic activity based purely on DFT calculations for those six parameters.
“Rather than you having to calculate the whole 28 descriptors, now you can calculate with only the six descriptors and get the methanol conversion rates you are interested in,” said Liu.
The team says they can also use the model to screen catalysts. If they can design a catalyst that improves the value of the six active descriptors, the model predicts a maximal methanol production rate.
Understanding mechanisms
When the team compared the predictions of their model with the experimental performance of their catalyst—and the performance of alloys of various metals with copper—the predictions matched up with the experimental findings. Comparisons of the ML approach with the previous method used to predict alloys’ performance showed the ML method to be far superior.
The data also revealed a lot of detail about how changes in energy barriers could affect the reaction mechanism. Of particular interest—and importance—was how different steps of the reaction work together. For example, the data showed that in some cases, lowering the energy barrier in the rate-limiting step alone would not by itself improve methanol production. But tweaking the energy barrier of a step earlier in the reaction network, while keeping the activation energy of the rate-limiting step within an ideal range, would increase methanol output.
“Our method gives us detailed information we might be able to use to design a catalyst that coordinates the interaction between these two steps well,” Liu said.
But Liu is most excited about the potential for applying such data-driven ML frameworks to more complicated reactions.
“We used the methanol reaction to demonstrate our method. But the way that it generates the database and how we train the ML model and how we interpolate the role of each descriptor’s function to determine the overall weight in terms of their importance—that can be applied to other reactions easily,” she said.
Discovery of a new catalyst for highly active and selective carbon dioxide hydrogenation to methanol
Wenjie Liao et al, Enhanced descriptor identification and mechanism understanding for catalytic activity using a data-driven framework: revealing the importance of interactions between elementary steps, Catalysis Science & Technology (2022). DOI: 10.1039/D2CY00284A
Provided by Brookhaven National Laboratory
citation: Machine learning framework IDs targets for improving catalysts (2022, May 10) retrieved 10 May 2022 from https://phys.org/news/2022-05-machine-framework-ids-catalysts.html
This document is subject to copyright. Apart from any fair dealing for the purpose of private study or research, no part may be reproduced without the written permission. The content is provided for information purposes only.